

최근 급여를 받은 척수성근위축증(Spinal muscular atrophy, SMA)의 치료제와 그 관련된 이야기를 해볼까 합니다.
미리 말씀 드리지만 병의 이해를 돕기 위해 쓰이는 단어가 조금 어려울 수 있습니다. 최대한 쉽게 풀어서 설명 드리겠습니다.
척수성근위축증은 상염색체 열성유전을 하는 유전적인 질환으로, 증상은 발생하는 시기에 따라 다소 다르지만, 대체로 사지근력의 저하가 있으면서 영아 혹은 신생아의 경우에는 호흡곤란이 함께 나타납니다. 증상 정도와 발생시점에 따라 아래 표와 같이 분류하게 됩니다.
| SMA타입 | 발생나이 | 최고 기능 | 사망시점 |
| 0 | 주산기 | 인공호흡기 필요 | 1개월 미만 |
| 1 | 0-6개월 | 혼자서 앉을 수 없음 | 2세 미만 |
| 2 | 18개월 미만 | 혼자서 서 있을 수 없음 | 2세 이상 |
| 3 | 18개월 이상 | 혼자서 서 있을 수 있음 | 성인 |
| 3a | 18개월-3세 | 혼자서 서 있을 수 있음 | 성인 |
| 3b | 3세 이상 | 혼자서 서 있을 수 있음 | 성인 |
| 4 | 21세 이상 | 혼자서 서 있을 수 있음 | 성인 |
병인으로는 우리몸에서 운동신경의 기능을 유지하는 단백질 중에서 하나인 survival of motor neurons(SMN)이라는 것이 있는데, 이 단백질을 표현하는 유전자의 이상(SMN1 동형접합 유전자결손(homozygous deletion);95%, 이형접합 유전자결손(heterozygous deletion);5%, 점돌연변이(point mutation);1%)으로 우리몸에 필요한 양을 만들지 못하여 증상이 발생하는 것으로 알려져 있습니다. 재미 있게도, 우리몸에는 특히 사람의 경우에는 SMN1유전자와 거의 같은 SMN2 유전자가 있으며, 환자에게도 이 유전자의 반복 수는 다소 남아 있는 상태입니다.
다만 SMN1유전자와 SMN2유전자 사이에는 구조적으로 사소한 차이가 있습니다. SMN2 유전자에서는 염기서열이 SMN1 유전자와 비교하여 한개가 바뀌어 있는데, 그 결과 SMN2 유전자에 의해서 약 85~90%가 7번 엑손의 내용이 비어있는, 즉 제 기능을 못하는 단백질이 만들어 집니다.
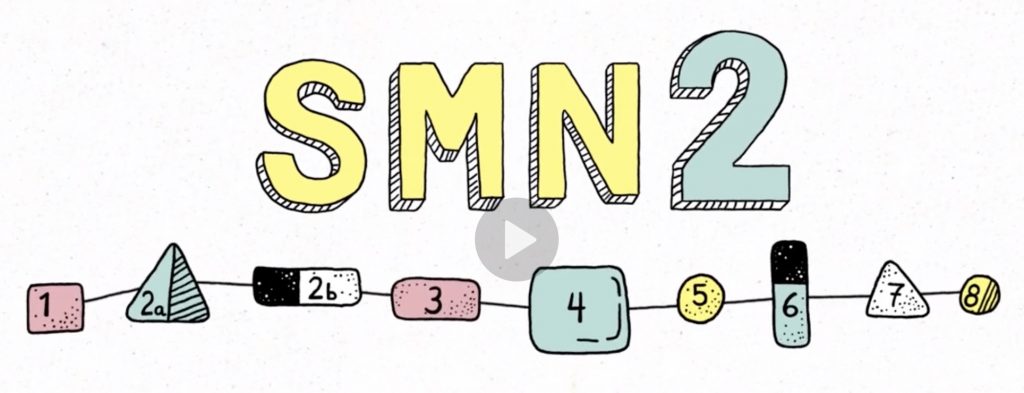
최근 치료법의 발달에 따라 유전자 치료 기법이 소개되었으며, 2019년 3월 우리나라에서도 급여를 받게 되어, 현실적으로 치료를 받을 수 있는 스핀라자(Spinraza, Nusinersen)에 대해 간단히 알려드립니다.
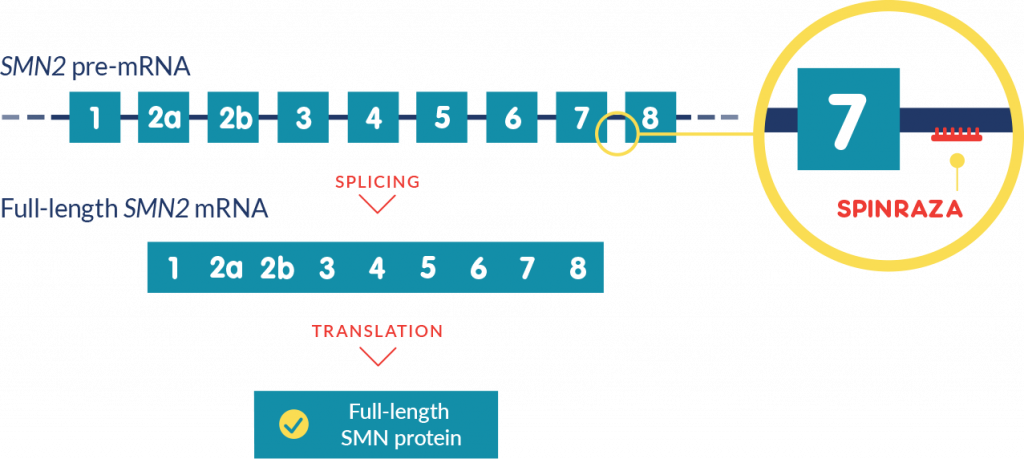
위에서 언급했듯이 SMN2 유전자에 의해 생성되는 단백질은 7번 엑손의 내용이 없는데, 스핀라자는 7번엑손 뒤에 작용하여 전체 길이의 mRNA가 생성되도록 하며, 최종적으로 SMN1 유전자에 생성되는 단백질과 같은 전체길이를 모두 갖는 단백질을 만들게 하여 증상을 개선합니다.
스핀라자는 다음 급여 조건을 모두 만족하여야 하며, 유지 용법 시에는 운동 기능의 유지 및 개선이 되어야 계속 쓸 수 있습니다. 그리고, 언제 치료를 중단하여야 한다는 것은 아직 기준이 없습니다.
- SMN1유전자의 결손 또는 변이 확인
- 만3세 이하 때에 척수성근위축증의 임상 증상과 징후 발현
- 영구적 인공호흡기를 사용하고 있지 않은 경우
주사제는 일반적으로 생각하는 정주 혹은 먹는 약이 아니고 경막내 투여하여야 합니다. 즉, 척수액 검사와 같이 허리에 천자를 하고 척수액을 주사액 용량만큼 제거한 후, 주사액을 넣어야 합니다.
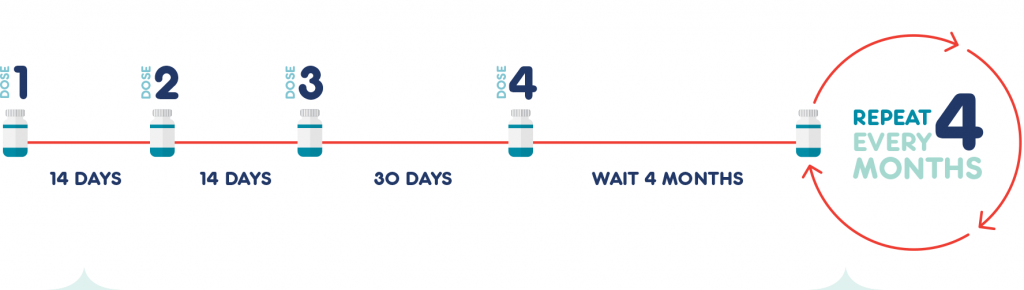
그리고 주사액의 스케줄은 다음과 같습니다. 주사약을 시작하고 2주간격으로 2차,3차를 시행하여야 하며, 한달후 4차, 그리고 4개월 마다 유지 용법을 해야 합니다. 그래서 첫해에는 총 6번 주사를 시행하게 됩니다.
그리고, 약제 투여전, 4회 투여(도입투여)이후, 5회 투여전(처음 유지 투여), 매 유지투여 이전에 임상적으로 평가를 하게 되며, 2회 이상, 현 상태 유지 혹은 개선이 되지 않으면 급여가 중지됩니다.
지금까지 진단을 받고, 치료가 없는 병이 신경근육질환에서는 굉장히 많았는데, 현실적으로 환자가 도움을 받을 수 있는 길이 열려 굉장히 고무적으로 생각하고 있습니다.
참고자료
1. https://spinraza-hcp.com
2. Kolb, S. J. and J. T. Kissel (2011). “Spinal muscular atrophy: a timely review.” Arch Neurol 68(8): 979-984.





